5.4 EMP_decostand
In multi-omics data analysis, the differences in dimension and magnitude of different data may lead to bias in analysis results. Standardized methods are therefore needed to eliminate these differences. Module EMP_decostand inherits the module decostand of vegan package, which can easily normalize data using logarithmic ratio, relative value, central logarithmic ratio, etc.
Note:
In the module
In the module
EMP_decostand, the parameter bySample can specify whether to standardize by sample or by feature. If no specific setting is provided, the parameter bySample will follow the recommendations in the vegan package.
5.4.1 Standardization of microbiome data
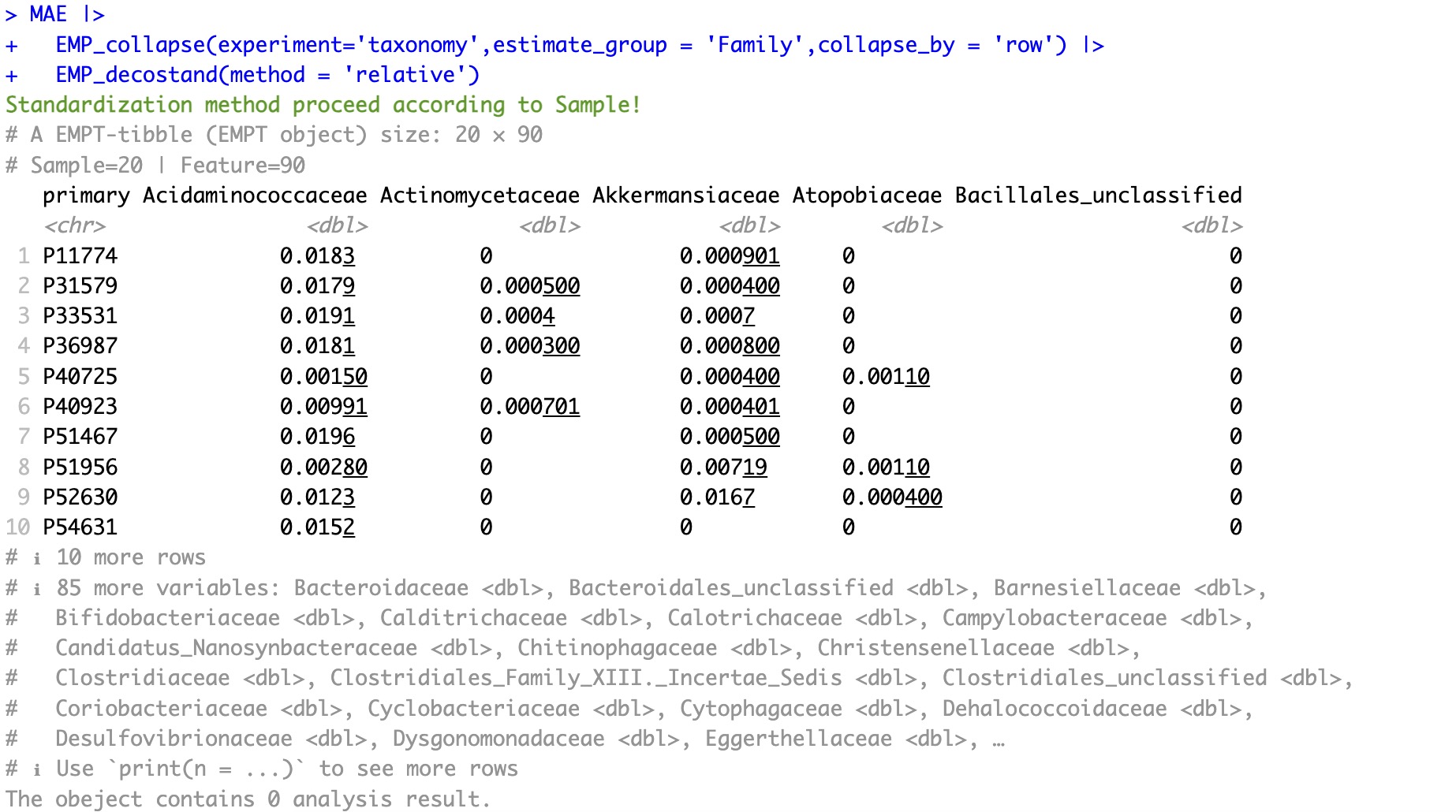
🏷️Example: Extract the assay of taxonomy and collapse it according to the Family column of rowdata, and convert the absolute abundance of the features of the collapsed assay into relative abundance.
MAE |>
EMP_collapse(experiment='taxonomy',estimate_group = 'Family',collapse_by = 'row') |>
EMP_decostand(method = 'relative')
5.4.3 Standardization of genomic data
🏷️Example: Extract the assay of host_gene and perform log conversion.
Note:
①When log, pa, and integer normalization methods are used, there is no need to consider the normalization direction.
②The transformation only applies a logarithmic change to values greater than 0, while values equal to 0 remain unchanged.
①When log, pa, and integer normalization methods are used, there is no need to consider the normalization direction.
②The transformation only applies a logarithmic change to values greater than 0, while values equal to 0 remain unchanged.
MAE |>
EMP_decostand(experiment = 'host_gene',method = 'log',logbase = 2)

Note:
The method can be written as log2+1, which means adding 1 to all data first, followed by a base-2 logarithmic transformation. If written as log or log+1, it will operate according to the parameter logbase.
The method can be written as log2+1, which means adding 1 to all data first, followed by a base-2 logarithmic transformation. If written as log or log+1, it will operate according to the parameter logbase.
MAE |>
EMP_decostand(experiment = 'host_gene',method = 'log+1')
